
線粒體在能量代謝和信號轉導等生物學過程中發揮著重要作用,是真核細胞不可或缺的細胞器。線粒體DNA耗竭綜合征(Mitochondrial DNA Depletion Syndrome,簡稱MTDPS)是一組罕見的遺傳疾病,由參與線粒體DNA(mtDNA)複製和維持的多個基因缺陷所引起,這些缺陷導致mtDNA編碼蛋白合成減少和氧化磷酸化(OXPHOS)不足,故而繼發各種代謝異常和多係統的異常臨床表現。
2023年12月20日,万博英超狼队网官方网 王陳繼團隊和上海市第一婦嬰保健院高昆團隊應邀在Trends in Molecular Medicine雜誌發表了題為Excessive BNIP3/BNIP3L-dependent mitophagy drives FBXL4 mutation-caused mitochondrial DNA depletion disease的綜述,概括了MTDPS 13亞型(MTDPS13)致病基因FBXL4突變導致線粒體自噬過度激活的內在分子機製,並討論了這種致命遺傳疾病的潛在治療策略。
MTDPS13相關症狀廣泛,個體差異很大,最常見症狀包括張力減退、神經發育遲緩和乳酸血症等。具有高能量需求的器官,如大腦、心髒、肌肉和腎係統,通常會受到更嚴重的影響。患者預後通常較差,平均3歲時死亡率約為30%。MTDPS13致病基因FBXL4是一個Cullin 3家族E3泛素連接酶複合體的底物識別亞基。自2013年首次描述FBXL4基因致病突變以來,在112例常染色體隱性遺傳患者中共記錄了59種致病突變。與雙等位基因缺失突變的患者相比,雙等位錯義突變、錯義/缺失突變複合雜合性患者往往生命更長。這一觀察結果意味著錯義變體可能在這些個體中發揮亞等位基因效應。
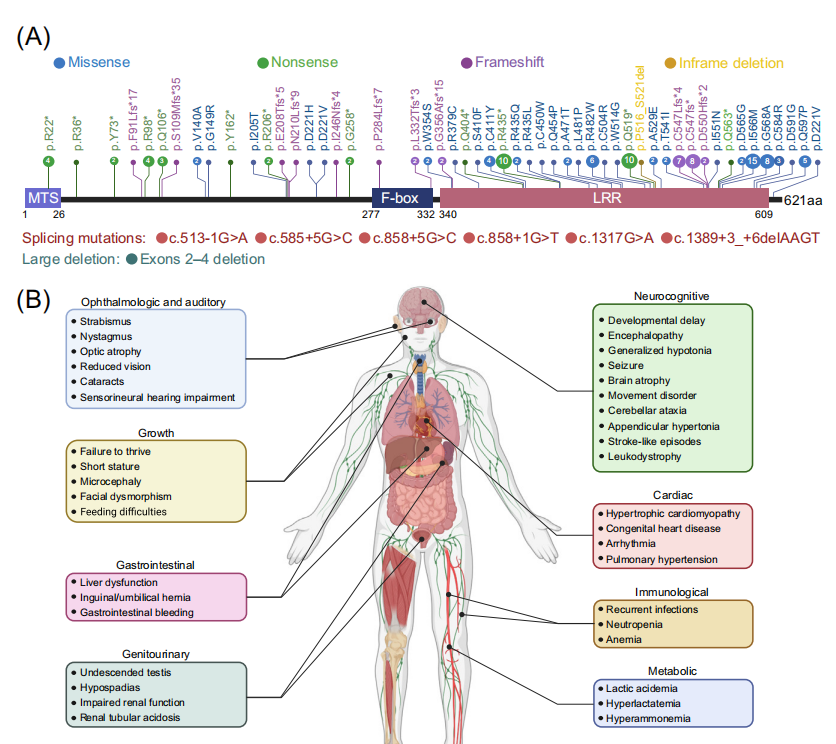
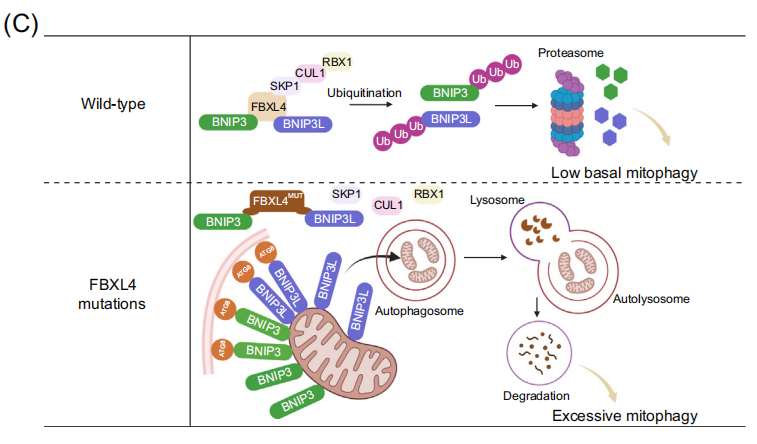
2023年發表的多項研究在該病的分子機理方麵取得重大進展,闡明了FBXL4通過控製線粒體外膜自噬受體BNIP3和BNIP3L來抑製基礎線粒體自噬(basal mitophagy)的關鍵作用。線粒體外膜蛋白BNIP3和BNIP3L具有LIR基序,能夠與自噬體ATG8家族蛋白相互作用,啟動線粒體自噬。FBXL4也定位於線粒體外膜,介導BNIP3/BNIP3L的泛素化-蛋白酶體途徑降解,使其維持本底低水平表達。而FBXL4缺乏則導致BNIP3/BNIP3L的大量積累,在培養的細胞係、MTDPS13患者的皮層神經元和FBXL4敲除/敲入小鼠模型中,FBXL4失活可觸發高水平的基礎線粒體自噬。FBXL4與其他外膜線粒體自噬受體,如PHB2、NIPSNAP1和FUNDC1沒有相互作用。此外FBXL4缺乏誘導的線粒體自噬不依賴於經典的Parkin-PINK1途徑。有趣的是,盡管FBXL4錯義突變位點集中在負責和底物識別的LRR結構域,但這些突變並不影響FBXL4與BNIP3/BNIP3L的相互作用,而使其與複合體其他亞基如Skp1、Cullin1和RBX1的相互作用顯著降低,出現活性CRL1FBXL4複合體的組裝障礙,顯著降低了對BNIP3/BNIP3L的泛素化降解作用。目前動態調控該過程的上遊信號事件仍在探索中。
至今MTDPS13仍無有效治愈手段,支持性的臨床幹預措施功效甚微。近期的基礎研究突破為未來探索該病的治療策略提供了希望和思路。比如,在基於腺病毒的基因治療方麵,可將野生型FBXL4 cDNA引入攜帶FBXL4基因突變的患者細胞中或使用CRISPR堿基編輯來校正基因組DNA中的FBXL4突變;而非病毒的方法則可使用ASOs或RNAi降低BNIP3/BNIP3L的mRNA水平,從而緩解患者細胞中BNIP3/BNIP3L蛋白的異常積累;化學小分子也有望成為潛在的治療策略,如設計PROTAC化合物靶向降解過量的BNIP3/BNIP3L;是否部分自噬抑製劑能夠減輕患者細胞中過度的基礎自噬也可以嚐試;通過PGC-1α激動劑促進患者細胞中的線粒體生物發生(Mitochondrial biogenesis)以平衡過度的線粒體自噬可能也是一個潛在的探索途徑。這些策略為MTDPS13未來治療方法的開發提供了可能的方向,但需要進一步在臨床轉化模型中對其有效性和安全性進行係統檢測和評估。
原文連接:https://doi.org/10.1016/j.molmed.2023.11.017